Using hydrogen as an energy carrier will potentially allow us to reduce our dependency on fossil fuels and to make the transition to a greenhouse gas neutral society. A major challenge involves the efficient generation of hydrogen from renewable sources. Solar energy stands out as a source with high potential, which could be used to generate hydrogen in a photoelectrochemical cell. We therefore pursue a detailed understanding of the semiconductor-water interface in order to provide a strong theoretical foundation for further technological advances. Another major challenge is, that appropriate materials for storing hydrogen are still missing, in particular for mobile applications. In the recent years a variety of different material classes has been proposed as potential candidates, including metal hydrides, carbon based nanostructures, metal-organic frameworks and doped polymers, to mention only some of them. Ab initio simulations allow to systematically investigate the microscopic mechanisms which drive the hydrogen sorption characteristics in these materials. We employ density functional theory to investigate the mechanisms of point-defect kinetics and their relevance for the hydrogen uptake and release in systems like in high-capacity hydrogen storage materials such as MgH,2, AlH3, NaAlH4 , LiBH4 and Li4BN3H10[1-7]. The goal of these studies is to provide interpretations for experimental observations and also to contribute to the design of better storage materials, i.e. to propose target-oriented engineering strategies to improve the control of the hydrogen sorption kinetics.
Our goal is to elucidate the physics and chemistry of hydrogen interactions with hydrogen storage materials by performing first-principles calculations based on density functional theory (DFT). By specifically addressing the behavior of hydrogen-related and other point defects, we are able to investigate the kinetics of hydrogen uptake and release, the decomposition of complex hydrides, and the effect of adding impurities to the system. Our approach takes into account that defects and impurities in non-metallic systems can occur in charge states other than the neutral state; this important aspect of the problem had not been addressed in previous computational studies performed to date, and our investigations show that it has extremely important consequences for defect concentration and diffusion. Other groups are now starting to apply the same methodology.
 |
|
Cluster of bonded Al atoms in an Aluminum hydride (AlH3) host crystal. The blue balls denote Al atoms and the white balls denote H atoms. |
. |
Point-defect-mediated dehydrogenation of Aluminum hydride
AlH3, one of the prime candidate materials for hydrogen storage applications, has been investigated in our group using hybrid-DFT simulations [1,2]. We show that under dehydrogenation conditions charged hydrogen vacancy point defects form in the AlH3 crystal, which have a strong tendency towards clustering. These clusters denote local nuclei of Al phase, and the growth of these nuclei eventually drives the AlH3/Al transformation, i.e., the dehydrogenation reaction of AlH3. The findings suggest that manipulating vacancy point defect concentrations would allow to control the dehydrogenation kinetics, opening up new engineering strategies.
Hydrogen-vacancy interaction in Mg and Al
Besides the dehydrogenation we also the study the hydrogenation process [4]. For the later the presence of vacancies in the bulk of the host metal and their interactions with atomic hydrogen is expected to play an important role. In a comparative study we have recently invetsigated the hydrogen-vacancy interaction in hcp-Mg and fcc-Al. Interestingly, a single V can in principle host up to 9 H atoms in Mg and 10 in Al. Evaluating the concentration of the H-V complexes for different H loading conditions reveals also that strong differences between Mg and Al. In the case of Al, up to 15 % of hydrogen atoms are trapped in single Vs even for very low H pressures, which slows down the diffusion of H atoms. In the case of Mg, these trapping effects are negligible for low H pressures. However, vacancies containing multiple H atoms and H-induced superabundant vacancy formation occur in Mg at much lower H loading pressures (about 1 GPa) than in Al (about 10 GPa)
| |
|
| |
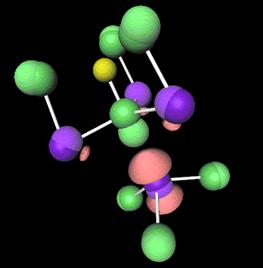
Fig. 2: Schematic representation of Li4BN3H10- a promising material for new forms of hydrogen storage. |
References:
[1] L. Ismer, A. Janotti, C.G. Van de Walle, Appl. Phys. Lett. 97, 201902
[2] L. Ismer, A. Janotti, and C.G. Van de Walle, Journal of Alloys and Compounds, In Press.
[3] M.S. Park, A. Janotti, and C.G. Van de Walle, Physical Review B 80, 064102 (2009).
[4] L. Ismer, M.S. Park, A. Janotti and C. G. Van de Walle, Phys. Rev. B 80, 184110 (2009)
[5] G.B. Wilson-Short, A. Janotti, K. Hoang, A. Peles, and C.G. Van de Walle, Physical Review B 80, 224102 (2009)
[6] K. Hoang and C.G. Van de Walle, Physical Review B 80, 214109 (2009)
[7] C.G. Van de Walle, A. Peles, A. Janotti, G.B. Wilson-Short, Physica B – Condensed Matter 404, 793 (2009) |
|